-
Large π-Conjugated Chromophores Derived from Tetrathiafulvalene
H. Jia, J. Ding, A. Hauser, S. Decurtins and S.-X. Liu
Asian Journal of Organic Chemistry, 3 (2) (2014), p198-202


DOI:10.1002/ajoc.201300144 | unige:34423 | Abstract | Article HTML | Article PDF
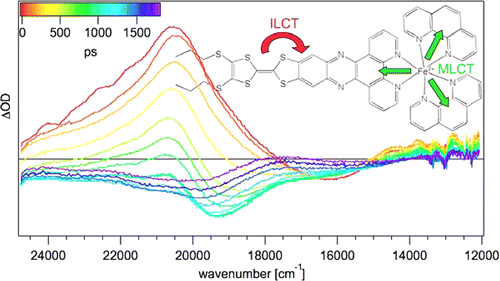
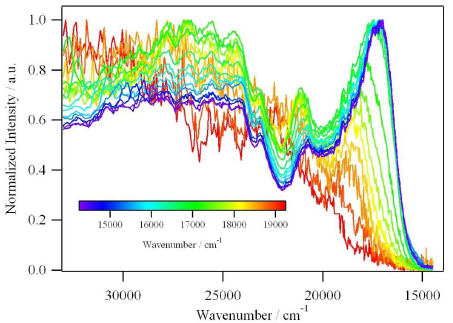
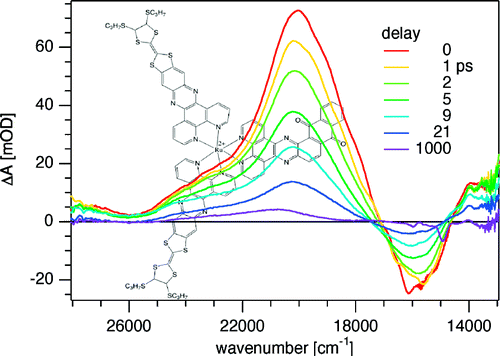
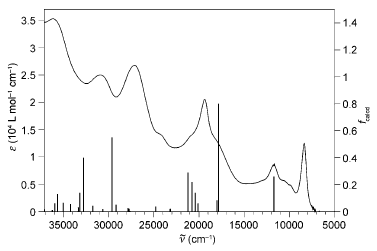
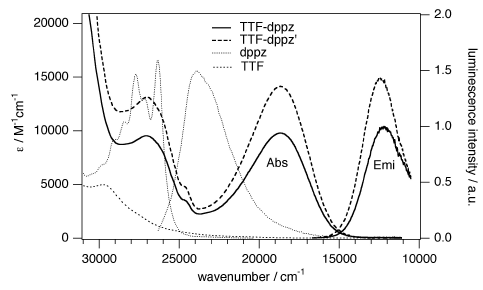
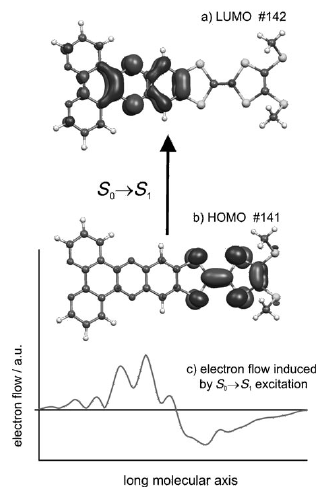
A large pi-conjugated chromophore composed of two dipyrido[3,2-a:2â,3â-c]phenazine (dppz) units directly fused to the central tetrathiafulvalene (TTF) core, has been prepared as a bridging ligand, and its strong binding ability to Ru2+ forming a new dinuclear complex is presented. The electronic absorption and luminescence and the electrochemical behaviour of the free ligand as well as the Ru2+ complex have been investigated in detail. The free ligand shows a very strong band in the UV region consistent with ligand centred Ï-Ï* transitions and an intense broad band in the visible region corresponding to an intramolecular charge transfer (ILCT) transition. Upon coordination, a metal-to-ligand charge transfer (MLCT) appears at 22520 cm-1 while the ILCT band is bathochromically shifted by 1620 cm-1. These electrochemically amphoteric chromophores have also been characterized by spectroelectrochemical methods. The oxidized radical species of the free ligand show a strong tendency to undergo aggregation, in which long-distance attractive interactions overcome the electrostatic repulsion. Moreover, these two new chromophores reveal an ILCT fluorescence with large solvent-dependent Stokes shifts and quantum efficiencies of 0.052 for the free ligand and 0.016 for its dinuclear Ru2+ complex in CH2Cl2.